La sindrome di Pfeiffer: cause, sintomi, trattamento
La sindrome di Pfayffer è estremamente rarauna malattia genetica che si verifica in media su uno su 100.000 neonati. Altrettanto spesso colpisce ragazzi e ragazze. Il sintomo principale della malattia è la coalescenza precoce delle ossa del cranio nel processo di embriogenesi, a causa della quale il cervello non può più svilupparsi normalmente.
Informazioni generali
La malattia è stata scoperta dal famoso tedescogenetista Rudolf Pfeiffer. Nel 1964 descrisse i sintomi di una malattia precedentemente sconosciuta, i cui principali segni erano anomalie nella struttura del cranio e fusione delle dita sui piedi o sulle mani (sindattilia). La sindrome è stata osservata in otto pazienti della stessa famiglia, che hanno indicato la natura genetica della malattia. Ulteriori studi hanno dimostrato che la sindrome di Pfeiffer è trasmessa da un tipo autosomico dominante.
Gli ultrasuoni possono rilevare questa malattia nel feto già nel secondo trimestre di gravidanza. Sull'ultrasuono si osservano anomalie dello sviluppo del cranio, degli organi interni e delle estremità.
sintomi
Nel 1993, il genetista americano Michael CohenPropose di classificare la sindrome di Pfeiffer in tre tipi a seconda della gravità dei sintomi. Ora la sua classificazione è generalmente accettata e ampiamente utilizzata nella letteratura medica.
I sintomi della sindrome di Pfayffer tipo 1 includonoCraniosinostosi - una condizione in cui una o più suture craniche si uniscono prematuramente formando un osso olistico. Di conseguenza, si notano cambiamenti nella forma del cranio e caratteristiche facciali anormali - ipertelorismo, orecchie basse. Ci può essere un peggioramento della vista, aumento della pressione intracranica, palatoschisi. Nel 50% dei pazienti si nota la perdita dell'udito. La maggior parte delle persone con questo tipo di malattia ha un livello normale di intelligenza e non ha anormalità neurologiche.
Nei bambini con sindrome di Pfayffer di tipo 1, esternai segni, come le dita larghe e corte su braccia e gambe, si osservano quasi sempre. Sindattilia è un sintomo possibile, ma non necessario, per questo tipo di patologia.

Questo tipo di sindrome è ereditariamente autosomica dominante.
Di seguito una foto della sindrome di Pfeiffer. L'aspettativa di vita in Russia per le persone che soffrono di questa malattia supera i 60 anni.

Per la malattia di tipo 2, il cranio è caratterizzato da aforma di "trifoglio", la fusione delle vertebre, la sindattilia e lo spostamento dei bulbi oculari in avanti (proptosi). Dai primi giorni di vita compaiono gravi disturbi neurologici.
Il terzo tipo di malattia è caratterizzato ancora di piùcraniosinostosi precoce. Il cranio è anormalmente allungato e non forma un "trifoglio". Vi sono anomalie di denti, ipertelorismo, ritardo mentale, malformazioni degli organi interni.
Durata al 2 ° e 3 ° tipola malattia varia da alcuni giorni a diversi mesi - i difetti degli organi interni e dei disturbi neurologici non sono compatibili con la vita. Le mutazioni responsabili di questi due tipi di sindrome di Pfeiffer si verificano sporadicamente e non sono ereditate geneticamente.
motivi
La sindrome di Pfayffer è associata a mutazioni del recettore 1fattore di crescita dei fibroblasti (FGFR1) sul cromosoma 8 o di crescita dei fibroblasti recettore del fattore di 2 (FGFR2) sul cromosoma 10. I geni che influenzano la mutazione responsabile per la normale crescita dei fibroblasti, che svolge un ruolo chiave nello sviluppo delle ossa del corpo.

trattamento
La medicina moderna non consente ancora di eliminaremutazioni nel genoma umano, anche se è noto in quali geni ci sia stata la perestroika. Pertanto, per la sindrome di Pfeiffer, come per la maggior parte delle altre malattie genetiche, il trattamento è sintomatico. Per i pazienti con tipo 2 e tipo 3, la prognosi è sfavorevole: numerose anomalie dello sviluppo non possono essere corrette né a livello medico né chirurgico.
La sindrome di Pfayffer di tipo 1 è compatibile con la vita. Il sintomo principale della malattia - la fusione prematura delle ossa del cranio - può essere corretto chirurgicamente. L'operazione è raccomandata per i bambini fino all'età di tre mesi.
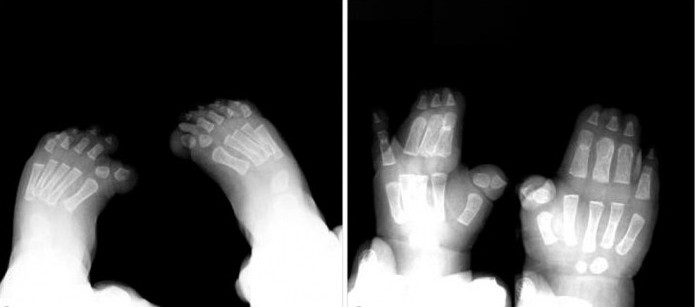
Inoltre, l'intervento chirurgico è efficace nel caso della sindattilia semplice. Il trattamento è meglio iniziare all'età di 18-24 mesi, durante la crescita attiva delle dita delle mani e dei piedi.